 醫學研究部(Department
of Medical Research)
醫學研究部(Department
of Medical Research)共 同 研 究 室 電 子 報
第五十四期 JUN.10.2018
本期目錄
下期主題
快速連結
隨著端午佳節來臨,時序也正式步入盛夏,酷暑炎熱的天氣應也不影響同仁對研究真知的追求。本期電子報將延續第49期次世代定序技術(NGS)的主題詳細說明如何使用「基因集(gene set)」來剖析與解讀高通量實驗數據。下一期電子報將介紹多因子檢測與磁性細胞分選應用新知,敬請期待,並竭誠歡迎您訂閱共同研究室電子報以收取儀器介紹、研究新知、與每月訓練課程資訊,更歡迎您與我們聯絡,給予我們建議與鼓勵。
網路預約系統的流式細胞分析儀
(LSRII)委託操作部份已升級上線。委託服務除了提供一般多色分析外,另可分析cell
cycle、side population、apoptosis及ROS變化。有相關需求者可在網路預約系統上即時預約。不分院內外,預約e指通,歡迎多加使用。已在線上申請委託者,請定期至「我的預約紀錄」查詢最新預約狀況。「臺大醫學校區學術演講及研討會公告平台」提供各單位舉辦之演講及
研討會公告園地,歡迎同仁查詢使用,並踴躍申請帳號發佈訊息。希望我們所提供的設備對您的研究有所助益,服務品質也令您滿意,為了共研長期的經營運作,請您於發表文章時惠予致謝共同研究室,作為服務成效評鑑之用。
TOP
許家郎專案助理研究員/醫學研究部
高通量實驗技術,可同時觀察或量測上百個,甚至上千至上萬個基因,雖然提供系統與全面性的數據,但如何解讀數據,卻成了另一個挑戰。本期將介紹如何使用「基因集(gene set)」來剖析與解讀高通量實驗數據。
基因集(gene-set): 一群具有某種關聯性的基因
基因集(gene set)指的是一群基因的集合,且這群基因具有類似的性質或功能。對於生醫學者最熟悉的基因集,莫過於生物路徑(biological pathway),例如IGF signaling pathway,一個反應路徑是由一連串基因間的活化、抑制、或交互作用所組成,如果把所有參與這條生物路徑的基因集合起來,就可稱做基因集。目前線上已有許多生物路徑的資料庫,例如由京都大學Minoru Kanehisa教授於1995年創立的KEGG(Kyoto Encyclopedia of Genes and Genomes)[1],與由歐洲生物資訊研究所(EBI)建構的Reactome資料庫[2]。這類型資料庫比較嚴謹,大多利用人工閱讀文獻,且兩基因間的調控關係需有明確實驗證據才會收錄,因此是很好的基因集來源,並且這些生物路徑資料庫通常會提供生物路徑「圖」,來呈現完整的調控關係,對於理解基因間的調控關係有很大的幫助。
另一個常見的基因集來自於Gene Ontology (GO) Consortium[3]。GO主要目的是訂定一套標準詞彙(controlled vocabulary),用來描述基因的功能。主要有三大分支,分別是Cellular component (CC),Molecular function (MF)與Biological process (BP)。從字面上來看,CC中的詞(term)是用來描述指基因產物在細胞內外的位置,例如plasma membrane;MF則用來定義基因產物分子層次的功能,例如DNA binding;而BP是用來描述基因產物所參與的生物路徑或機制,例如cell death。當有了標準詞彙,接下來就是把這些詞指派給各個基因,而指派的方法可分為人工閱讀文獻收集與計算預測基因功能這兩種策略。所以當你在一些基因或蛋白質資料庫(例如Entrez Gene或UniProt),可以看到指派給該基因的GO後面,會有一組證據碼(evidence code),來告訴你是依據什麼方式將這個詞分配給該基因,如果是從文獻得到,還會附上PubMed ID。有了基因與詞的關係後,就可以將每個詞相對應的基因收集起來,成為基因集。需要特別注意,GO的詞之間,有些是有繼承關係,因此如果把這些詞的關係建構出來,會形成一個類似樹狀結構(如圖1),用靠近根部(root)的詞,定義越粗略,越接近枝葉的詞則越精準。所以當我們在收集基因集時,針對某個詞,不是只把賦與該詞的基因收錄進來,而連有繼承該詞的基因也要收錄進來。以圖1中的DNA replication這個詞為例,如果基因A被指派這個詞,當你要收集DNA recombination這個詞的基因集時,也要把基因A收錄進來。
如一開始所提到,基因集是一群有某種關聯性的基因,因此不一定是功能上的相似或參與同樣的生物路徑,也有可能是代表某種表徵。以另一個知名軟體GSEA(稍後會特別介紹)所附屬的基因集資料庫MSigDB[4],除了收錄前面所提到的生物路徑和GO資料外,它們所收錄的基因集還包括基因坐落在染色體的位置是否相近,是否被同一個轉錄因子或microRNA所調控,甚至是否是某個特別藥物或癌症有關的signature等。
核心概念:enrichment
利用基因集解讀高通量資料的概念其實很簡單,就是去觀察我有興趣基因是否集中在某些基因集上,如果有興趣的基因都屬於某個基因集,那代表這個實驗資料跟這基因集所屬的功能或事件有關。但這就牽涉到,需要多少才算「多」?這時我們可以用統計或計算方法來回答這個問題。
基因集的分析策略可以分成兩類: over-representation analysis (ORA)與gene-set enrichment analysis (GSEA)(圖2)。這兩種方法最大的差別是,ORA會先經過篩選,挑出我們有興趣的基因,而GSEA則不經過篩選基因的動作。以轉錄體資料為例,實驗設計上,通常會比較兩種狀態,並利用統計方法找出哪些基因具有「表現差異」,可能會設定統計檢定的p值或fold-change,來決定這是我們有興趣的基因,接著就針對這群基因做解讀。這樣篩選的過程,p值或fold-change如何設定才能抓出真正具有「生物意義」的基因,且這種方法把每個基因都視為同等重要,然而每個基因的貢獻程度也許是不同的(即表現量差異大的可能比較重要)。而GSEA不做任何篩選動作,將所有實驗資料放入分析。
首先先介紹ORA的方法,我們可以用圖3A來說明。最外圍的圈,我們稱作背景集(background set),背景集通常是物種的所有基因或者實驗方法所量測到的所有基因。我們關心的是:有興趣的基因中(genes of interest),與某個基因集(gene set),共同基因有幾個(即圖3A中的k值),且需要有多少共同的基因(即k值要多大)才值得用此基因集來解讀我們的資料。看到這張圖,也許有人會聯想起在統計課時,常用的一個例子:一個籃子裡有N顆球,其中有m顆紅球,剩下皆白球,隨意抓取n顆球,其中k顆紅球的機率是多少?對於這類問題,我們可以用超幾何分布(Hypergeometric distribution)或二項式分佈(binomial distribution)來計算觀察值k的機率。同樣的,可以用這樣的方法來計算觀察到共同基因大於k的機率,如果機率很低,那代表這基因集與我們的資料有很強的關係,並非隨意都可以觀察到這樣的結果。我們也可以把圖3A轉變成圖3B的表格形式,這樣的列聯表 (contingency table),就可以採用費雪精確性檢定(Fisher’s exact test)或卡方檢定(Chi-square test)方法,來檢測基因集與我們有興趣的基因是否有相依性。
由前一段的介紹,可以看到ORA並沒有考慮到實驗數值,而把每個基因的貢獻視為相同,且分析前需決定那些基因才是我們有興趣的。這樣的做法有可能產生具有「統計意義」的基因集是由一些變化量不大的基因所組成,或者具有「生物意義」的基因,由於變化量不夠大,在一開始就被我們排除在外。GSEA則另一種分析策略,可以避免掉上述情況。GSEA的概念如他們在2005年在PNAS發表的圖所示(圖4)[5],首先將高通量實驗所量測到的基因排序,排列的順序是根據實驗量測到的數值決定。例如細胞加藥實驗,我們可以將有加藥與沒加藥分別做RNA-seq或microarray,並計算出所有基因在兩種狀態的fold-change或signal-to-noise等。下一步,將某個基因集內的基因,在經過排序的基因列表上標註出來。我們先想像一件事,假設生物路徑的活化與否是表現量有關,如果某藥物會影響某條生物路徑,那這條生物路徑的基因在經排序的基因列表呈現怎樣的分布呢?如果這藥物會活化此生物路徑,那我們期待這些基因都會坐落在起始的位置上(相對於未加藥,這些基因表現量變高);反之,如果是抑制,那會期待大部分會坐落在尾巴(相對於未加藥,這些基因表現量變低)。那要如何去「量化」這個現象呢?GSEA採用一個稱random walk的方法,也就是從基因列表的頭走到尾,如果碰到是基因集的基因就加分,不是則扣分。走完一趟後,回頭看走到哪兒時,分數最高(或最低),這個分數就是所謂的enrichment score (ES)。如果基因集都坐落在起頭,那會得到很高的正分;相反的,如果都坐落在尾巴,則會得到很小的負分。問題又來了,這樣的分數真的夠大(或夠小)嗎?會不會隨便挑幾個不相干的基因當基因集也會得到相同的分數?為了回答這個問題,GSEA利用permutation testing的方法,也就是隨機抓取同等數量的基因當基因集,並計算得到隨機的ES,去估算實際觀察到的ES的P值,如果P值小於所設定的統計條件,就可以確保這ES並不是隨機就會發生。除了由p值來判定外,還要回去檢查它所呈現的enrichment plot,確保尖峰或峰谷不是在中間位置,而是在所右兩側(圖4B和C)
基因集分析方法最大的優點是,不需要一個個去查文獻,來了解每個基因的功能,而可以很快速地知道,實驗資料可能跟那些生物路徑或功能有關聯,以利提出假說並設計下個實驗驗證。而缺點是這些基因間的關聯是什麼無法直接告訴你,仍需花精力解讀每個基因集。雖這篇文章以ORA和GSEA兩種方法來介紹,但大部分時候,這名稱已經被混用,甚至還有其他的名稱,例如pathway enrichment analysis、function enrichment analysis、GO enrichment analysis等等。無論是用什麼樣的名稱,概念都是如這裡所提到的,只是使用的基因集不同,或使用的統計方法不同。基因集的分析方法,在data-driven hypothesis的研究策略上,已經有許多成功的例子。下一次心中如果有某些新想法,試著先去網路上找找是否有類似的實驗資料,並利用這些方法來測試你的假設。
[1] http://www.genome.jp/kegg/
[3] http://www.geneontology.org/
[4] http://software.broadinstitute.org/gsea/msigdb/index.jsp
[5] https://doi.org/10.1073/pnas.0506580102
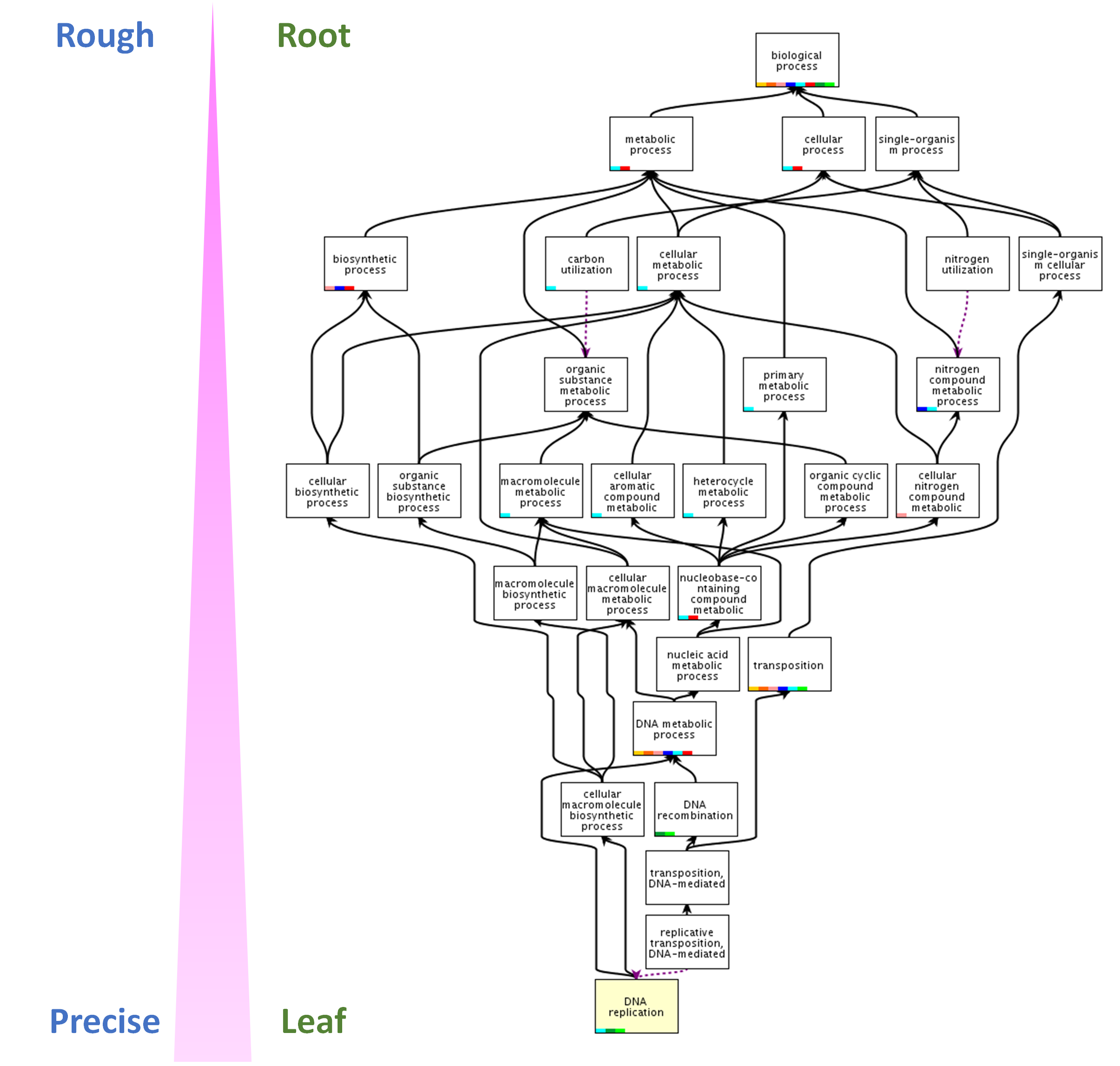
圖1、Gene Ontology (GO)詞間是具有繼承關係。越靠近根部的詞,定義越粗略,越靠近枝葉的詞,定義越精準。此例是以GO term “DNA replication”為起點,追溯回根部(biological process)所形成的結構。
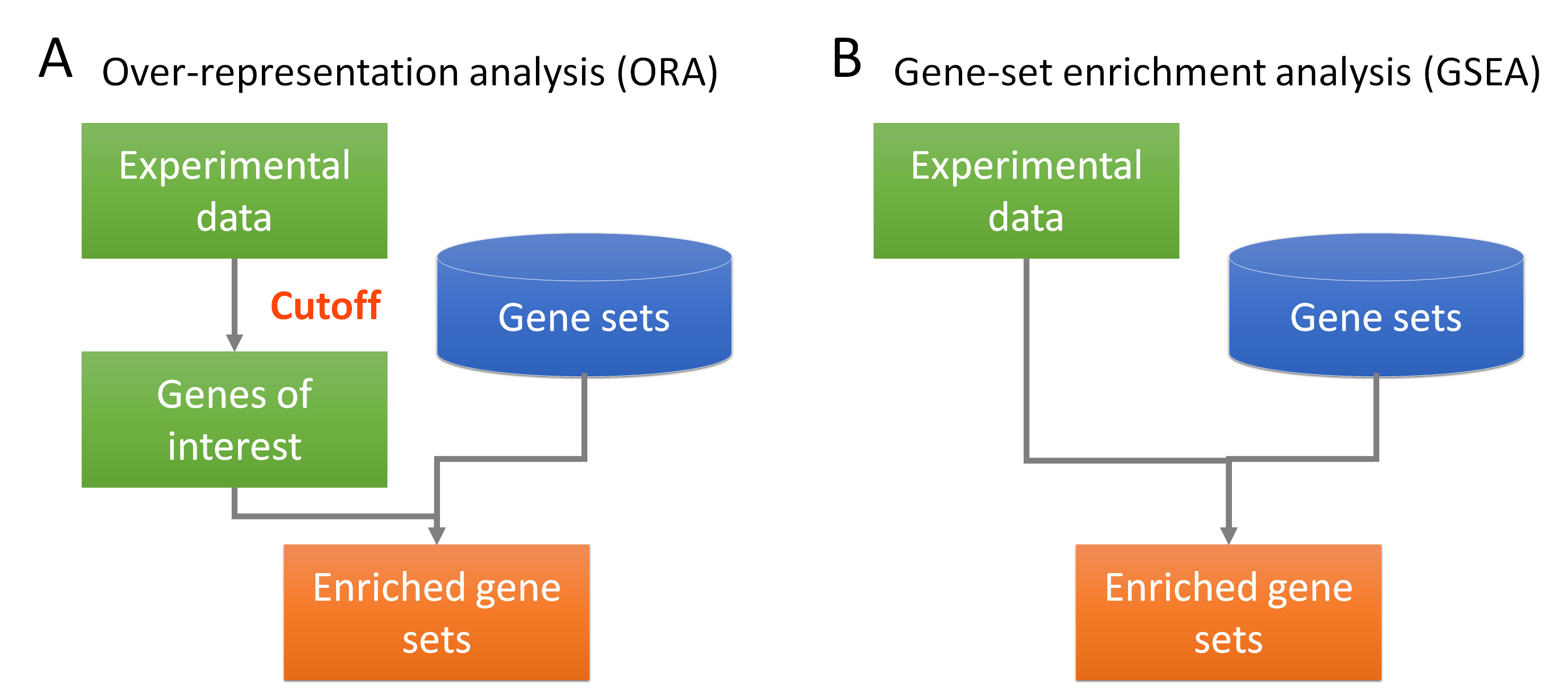
圖2、Over-representation analysis (ORA)(A)與gene-set enrichment analysis (GSEA) (B)流程上的差異。
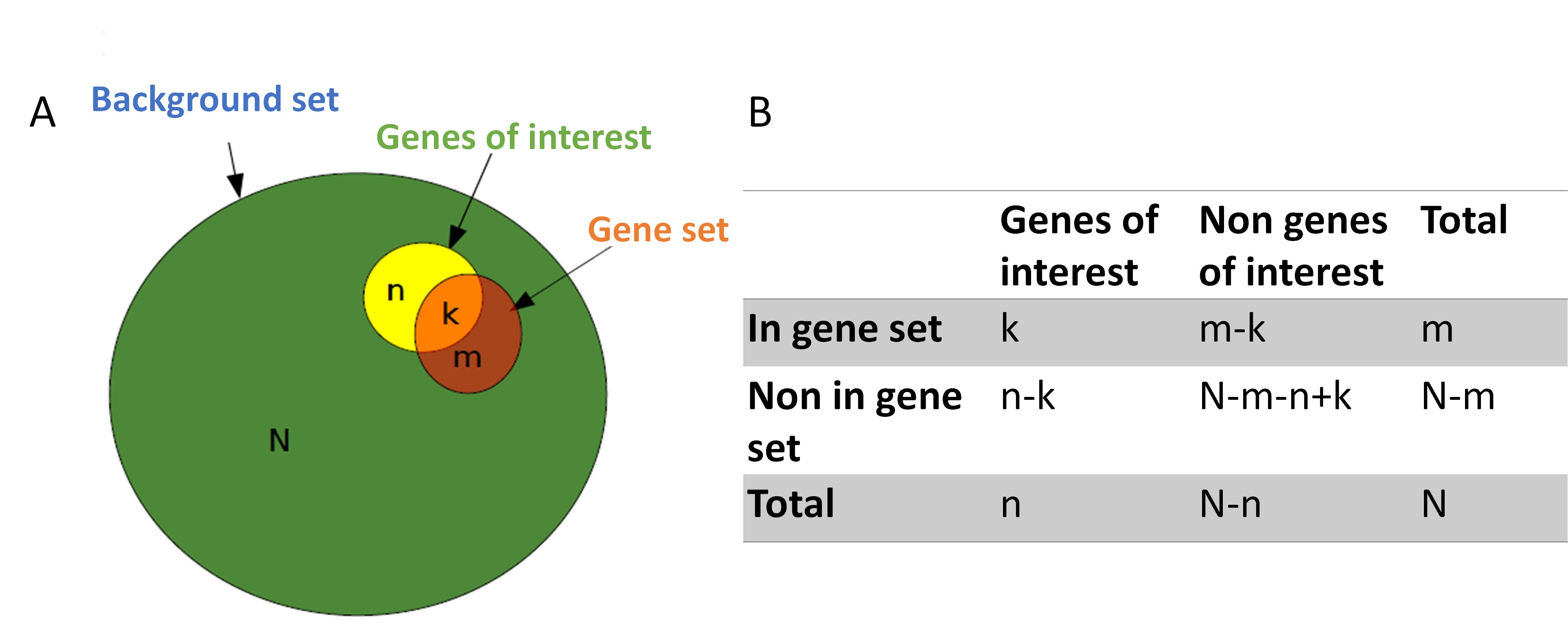
圖3、Over-representation analysis的概念就是比較有興趣的基因是否會被某基因集(Gene set)涵蓋。這個概念可以用Venn diagram的方式(A)或列聯表(contingency table)(B)來表示,就有相對應的統計檢定方法可以使用。
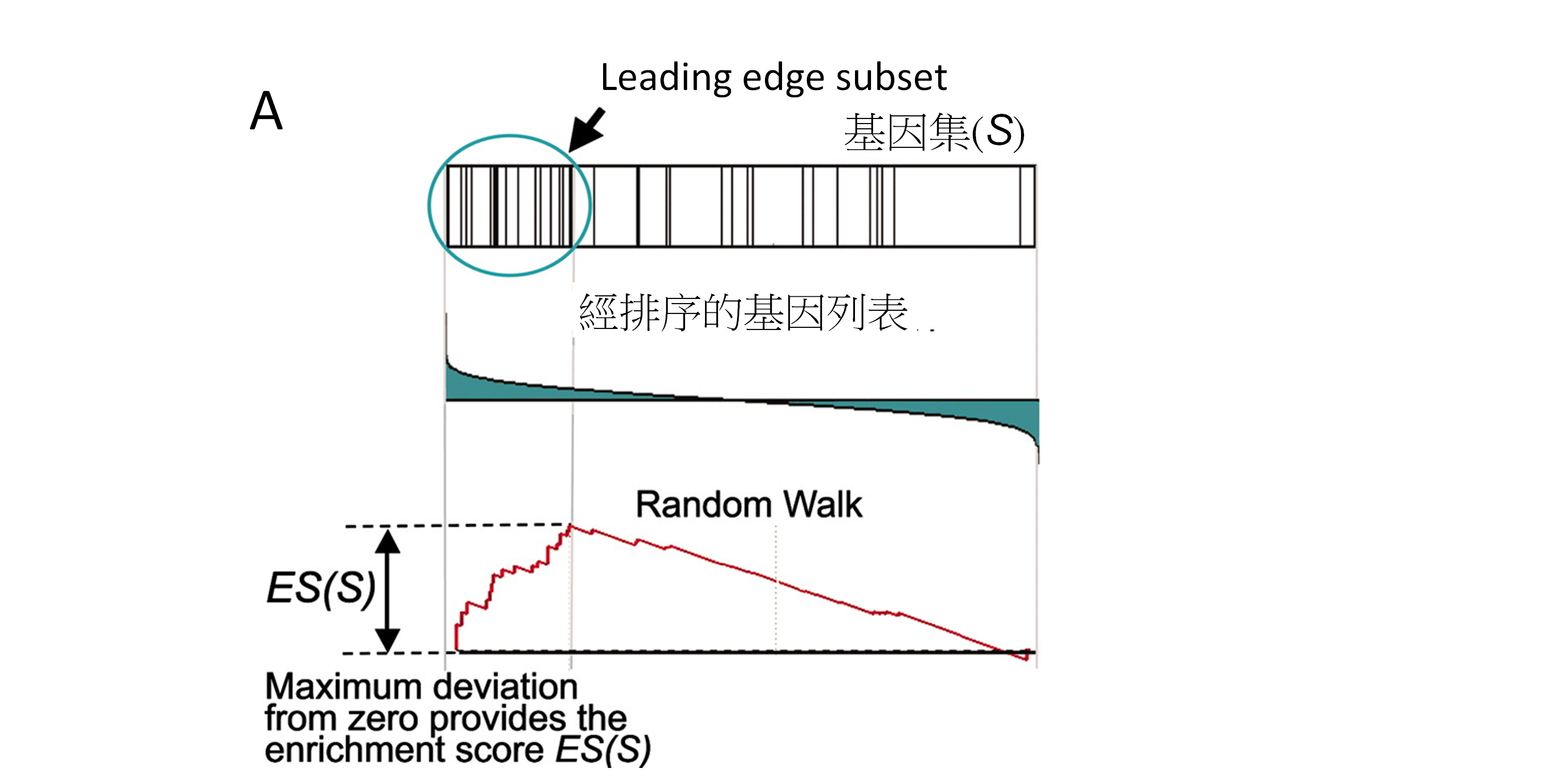
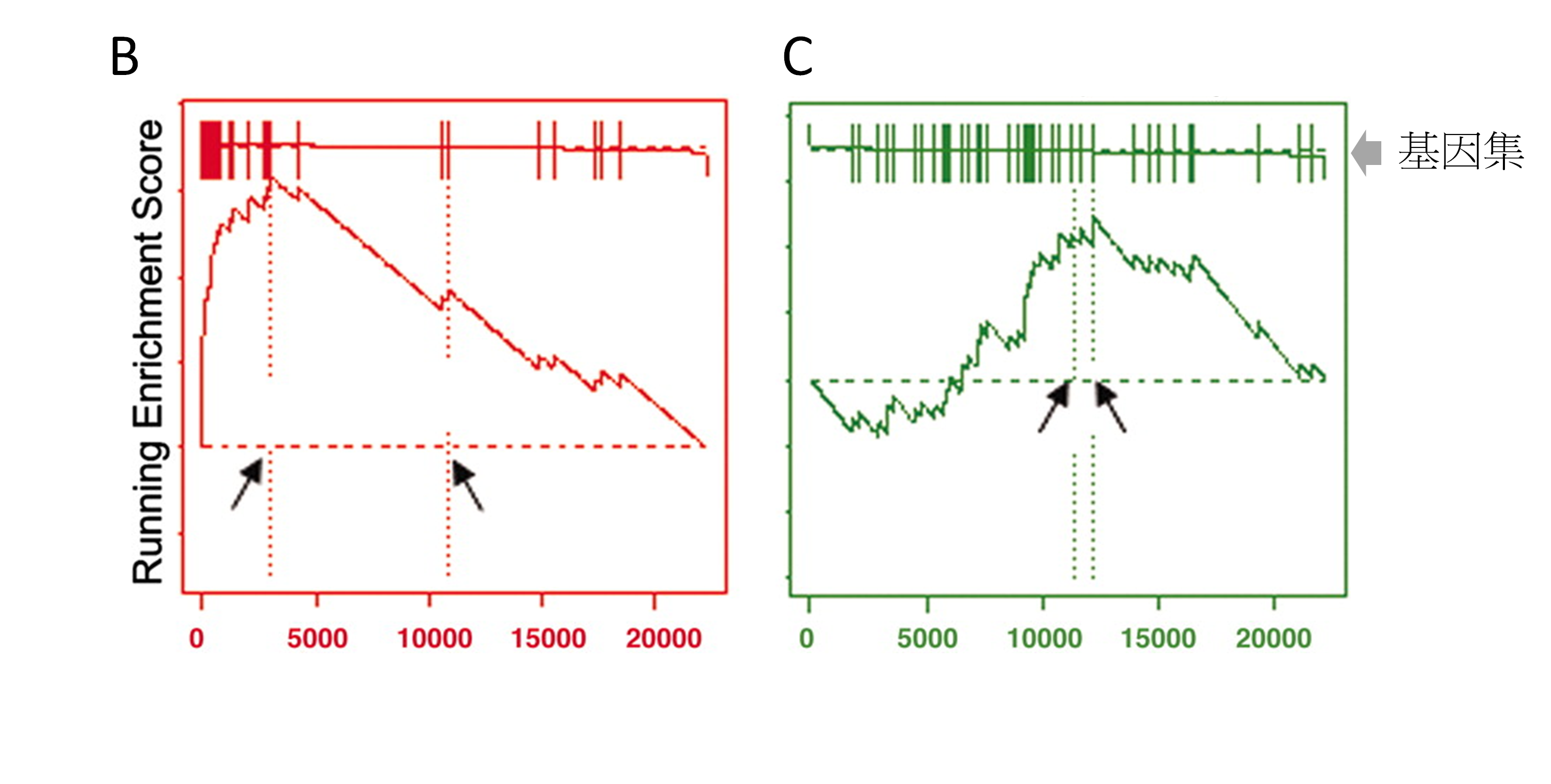
圖4、(A)
Gene-set enrichment analysis (GSEA)方法概念圖。(B)和(C)
enrichment plot,其中(B)的基因集分佈才是我們有興趣的結果。圖來自於Subramanian
A et al. (2005) PNAS. doi: 10.1073/pnas.0506580102。
TOP
儀器訓練課程:課程網路報名
| 06月12日 |
流式細胞儀(BD; Calibur)
|
|
| 06月20日 | 全波長微盤光譜分析儀(Multiscan GO) |
歡迎您訂閱共同研究室電子報以收取儀器訓練與研究新知課程講習相關資訊 。
為持續提供優質之研究服務,便於日後聘用專職技術人員、購置新儀器、現有儀器汰舊換新與維護保養等等,敬請於使用共同研究室資源並發表論文時,於論文致謝(Acknowledgement)處加入致謝共同研究室之文句,並於論文發表時通知共同研究室管理人員。致謝文句請依實際使用情形書寫,或請參考以下範例:We
thank the staff of the Core Labs, Department of Medical Research, National
Taiwan University Hospital for technical support.