共 同 研 究 室 電 子 報
第四十九期 JAN.10.2018
本期目錄
下期主題
快速連結
次世代定序技術 (NGS) 的問世,成為基因體研究的最佳利器,也成功應用在各類「體學 (Omics)」研究上。本期將著重在NGS於轉錄體的研究與應用。下一期電子報將介紹製藥用隔離裝置,敬請期待!並竭誠歡迎您訂閱共同研究室電子報以收取儀器介紹、研究新知、與每月訓練課程資訊。更歡迎您與我們聯絡,給予我們建議與鼓勵。
共同研究室全自動毛細管核酸分析儀(QSep100)網路預約系統擬於本月底上線!請留意網頁公告,歡迎多加使用。本月份「臺大醫學校區學術演講及研討會公告平台」將公告多場精彩演講及課程,歡迎同仁查詢使用,並踴躍申請帳號發佈訊息。希望我們所提供的設備對您的研究有所助益,服務品質也令您滿意,為了共研長期的經營運作,請您於發表文章時惠予致謝共同研究室,作為服務成效評鑑之用。
TOP
醫學研究部 許家郎專案助理研究員
次世代定序技術(Next-generation sequencing,簡稱NGS)的發展與問世,為基因體學研究,帶來革命性的影響。所謂「次世代」定序,是泛指有別於Sanger定序的方法,並具有通量高、速度快、與費用低等優勢。而利用不同的分子生物學與物理化學技術,將序列片段分離出來與建庫(library),再利用定序技術來辨識這些片段的核酸組成,就延伸出各種”-Seq”的資料,例如RNA-seq就是將RNA片段定序的研究技術,而exome-seq就是將基因組內exon的部分擷取出來並定序的技術,另外還有許多研究epigenomics的實驗方法,包括ChIP-seq、ATAC-seq、HiC-seq等等。這一期,將著重在NGS技術在轉錄體的應用,特別是RNA-seq。
如前所提,RNA-seq就是將RNA片段定序的研究方法。利用定序技術來研究轉錄體並非全新的概念,在Sanger定序的時代,就有將所有RNA片段建庫與定序的研究方法,例如EST(Expression Sequence Tag)與SAGE(Serial Analysis of Gene Expression),成功的應用在許多基因體計畫上,特別是在未知基因(novel gene)探索上。但是,Sanger定序的主要缺點,就是通量(throughput)低與速度慢,因此應用的範圍有限;而NGS高通量的特點,造就NGS可以成功的廣泛地應用於轉錄體研究上。由於建庫與定序是一種「隨機」抽樣的過程,無法確保每一個片段都可以成功定序出來。再加上,基因表現有高有低,高表現的基因,會有大量的RNA片段,因此在定序過程中,有較高的機率被抽中;反之,低表現的基因,由於RNA片段量少,因此被抽中的機率相對低。為了讓低表現的RNA有機會被抽中,那就必須增加抽樣的次數,也就是提高通量。藉由通量的提高,不但可涵蓋完整的RAN資訊,也可獲得準確的量化資料。
RNA-seq除了可用於量測基因表現量外,還可從RNA-seq資料獲取其他基因體或轉錄體資訊。在NGS技術中,我們把定序儀(sequencer)產出的序列片段,稱作read。藉由分析RNA-seq read,可取得的資訊與應用,詳列如下(圖1):
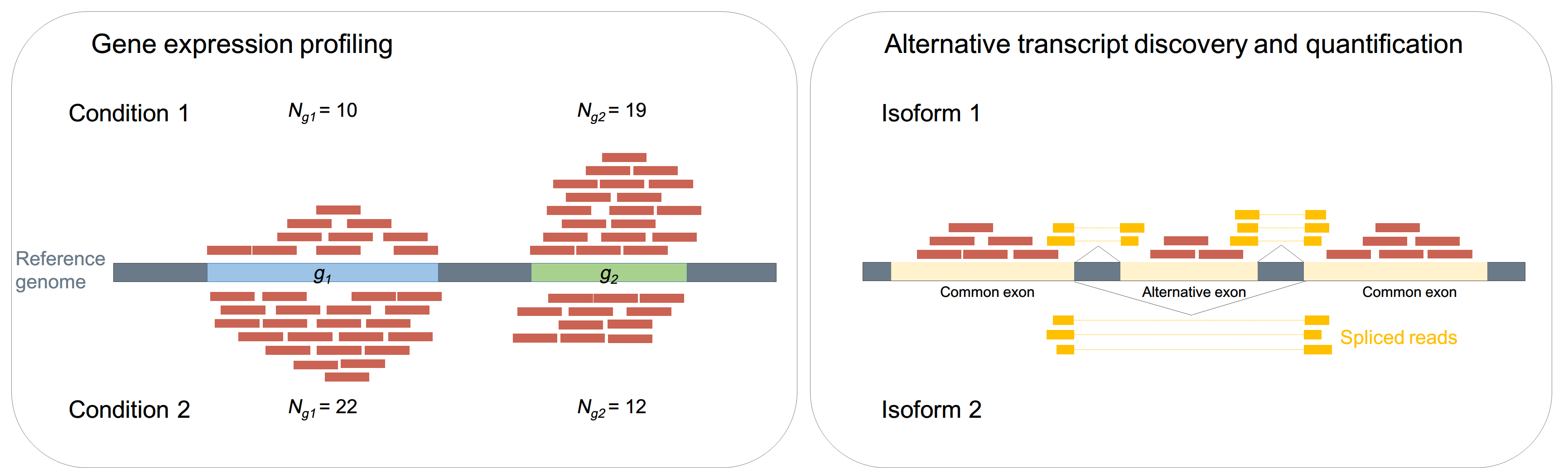

圖1、RNA-seq資料能取得的資訊。
■ Gene expression
這是RNA-seq最廣為人知的應用項目,概念是基因轉錄出來的RNA片段,與定序儀產出的read數成正比,因此可將來自於某基因序列數量(read count),來代表該基因的表現量。
■ Gene discovery
在基因體上,能夠被轉錄出來的區域,就能稱作廣義的「基因」。如果RNA-seq read比對到基因體參考序列(reference sequence)的區域,是從未註解過,則該區域將可能是新的基因區域(locus)。例如,lncRNA (long non-coding RNA)是一種與mRNA結構類似,但卻不具有轉譯出蛋白質能力的RNA。由於NGS的發展,有大量的lncRNA被發現。另外,如果研究的物種缺乏參考序列,也可以利用RNA-seq的方式來尋找具功能性的基因。
■ Alternative transcript discovery and quantification
在真核生物中,由於基因剪接(alternative splicing)機制,一個基因能產生一種形式以上的轉錄本(transcript)。可以藉由分析read是否橫跨不同的外顯子(exon)組合,來推論是否有新的轉錄本;同理,也可以藉由不同轉錄本上的read個數,來量化其不同轉錄本的表現量。
■ Gene fusion discovery
概念類似alternative transcripts,如果read有部分屬於A基因,另一部分屬於B基因,代表兩基因有fusion的可能性。雖然fusion主要是在基因體結構異常造成的,但是如果能發現其轉錄本,代表此fusion gene是具有功能性。
■ Gene variant identification
由於RNA是以基因體為模板所抄錄出來的,因此read上也帶有基因體資訊。當我們把read與參考序列做比對,就可以尋找出可能的變異點(variants)。然而,在轉錄過程中,RNA上的特定位置會受到編輯(editing),因此如何區分是RNA editing還是DNA variant,是後續資料分析中,需注意的地方。
■ Allele-specific expression
我們成對的體染色體中,一套來自於父親(paternal),另一套來自於母親(maternal),大部分的基因在兩套染色體中都會表現,但有少數的基因只會由某一套來表現,例如印痕基因(imprinted genes)。由於read上帶有基因變異點的資訊,如果該變異點是heterozygous,則可以藉由計算不同allele的read個數,來推論是否為allele-specific expressed gene。
然而,並非所有的RNA-seq資料都可以獲取這些資訊。依據建庫的方法,RNA-seq還可以分做whole transcriptome sequencing (WTS)與mRNA-seq。WTS通常除了rRNA外,其他RNA類型都可偵測到,包含mRNA與lncRNA;然而,mRNA-seq顧名思義,是針對mRNA做定序,因此在建庫前,先利用分子生物學方法,將mRNA序列抓取出來,最廣泛使用的策略,是利用探針(probe)針對mRNA 3’端的poly-A序列,將mRNA抓取下來。從這兩種建庫的方法,可以想像能獲取的資訊就有很大的差異:前者可以觀測大部分的RNA,但後者只能觀測mRNA。除建庫的方法不同會影響所得的資訊外,NGS實驗還牽涉到許多參數,不同的參數選擇也會影響所得的資訊範圍與準確性。
■ 序列長度(read length)
定序儀藉由設定cycle數,來決定序列長度。序列長度有幾個主要考量:首先,在資料分析上,會把read與參考序列做比對,如果read太短,容易產生單一read對應到多個區域(multiple-hits)。另外,在真核細胞中,由於有基因剪接的機制,如果read剛好橫跨不同外顯子(exon)的序列,在比對過程中,會把read切成兩段,才有辦法成功比對到參考序列上。當read切成兩段後,如果有一段片段太短時,將無法正確的對應到參考序列上。目前市面上常見的定序儀,長度都能夠達到50鹼基對(base pair)以上,因此比對時發生對應至多區域的機率,將大大降低。如果研究目的只是要量測基因表現量(gene expression profiling),序列長度50-75 bp就足夠;但如果是想尋找alternative splicing或者gene fusion這結構變化時,建議使用更長的序列長度。
■ 單端(single-end)與雙端(pair-end)序列
Single-end指只針對RNA片段一端定序,而pair-end指對RNA片段兩端定序,大部分的定序儀都能提供這兩種定序模式。在RNA-seq中,single-end就足以提供各種資訊,但如果研究目的是要尋找基因剪接或gene fusion,那建議使用pair-end。如果一組pair-end read,一端坐落在某個外顯子或基因上,一端坐落在另一個外顯子或基因上時,這種資訊將提高尋找基因剪接或gene fusion的可信度。
■ 序列深度(read depth)
在RNA-seq中,所探討的序列深度指的是:需要有多少read產出,才能提供足夠的資訊。序列深度,取決於RNA-seq實驗的目的。例如,WTS和mRNA-seq,前者就需要後者較大的深度,因為前者涵蓋的RNA種類與數量大於後者。如果同樣是WTS,但單純量測基因表現量,與想研究基因剪接或尋找fusion gene時,後者就需比前者更大的深度。一般來說mRNA-seq實驗,建議至少能達到1千萬條序列(10 million reads),而WTS至少要5千萬條序列。
NGS技術逐漸普及,價格也逐漸降低,不再是高不可攀的研究方法。但執行前,要釐清研究目的,並選擇正確的平台與參數,才能夠獲取有用的結果。醫學研究部新增了次世代基因定序與生物資訊分析服務,如有研究上的需要,歡迎與我們聯絡。
聯絡資訊:
許家郎博士 分機:
65989
延伸閱讀:
Byron S.A. et al. Nature Review Genetics, 2016; 17:257-271
TOP
儀器訓練課程:課程網路報名
| 01月22日 |
高階多色流式細胞儀(BD LSR II)原理介紹與實機操作
|
|
| 01月22日 | MagCore HF16磁珠核酸萃取儀 | |
| 01月22日 | 多功能流式微粒螢光分析儀(Labscan100/Luminex) | |
| 01月24日 |
螢光顯微鏡暨影像擷取系統(Nikon TS-100&DS-L2)
|
歡迎您訂閱共同研究室電子報以收取儀器訓練與研究新知課程講習相關資訊 。
為持續提供優質之研究服務,便於日後聘用專職技術人員、購置新儀器、現有儀器汰舊換新與維護保養等等,敬請於使用共同研究室資源並發表論文時,於論文致謝(Acknowledgement)處加入致謝共同研究室之文句,並於論文發表時通知共同研究室管理人員。致謝文句請依實際使用情形書寫,或請參考以下範例:We
thank the staff of the Core Labs, Department of Medical Research, National
Taiwan University Hospital for technical support.